Chemical reactions involve the breaking of existing bonds and the formation of new ones between atoms. That description sounds simple, but what does it mean microscopically? If enough energy is supplied to a molecule, its atoms begin to vibrate more strongly. If this motion becomes large enough, a bonded pair of atoms may separate to the point that the bond is effectively broken. After further thermal motion, the atoms can rearrange and form new bonds, producing a different molecular structure. This rearrangement is favorable when the resulting molecule, the product, has lower energy than the initial molecule, the reactant. Along the way, the system typically passes through a highest-energy configuration, where old bonds are being broken and new ones are only beginning to form. This configuration is known as the transition state.
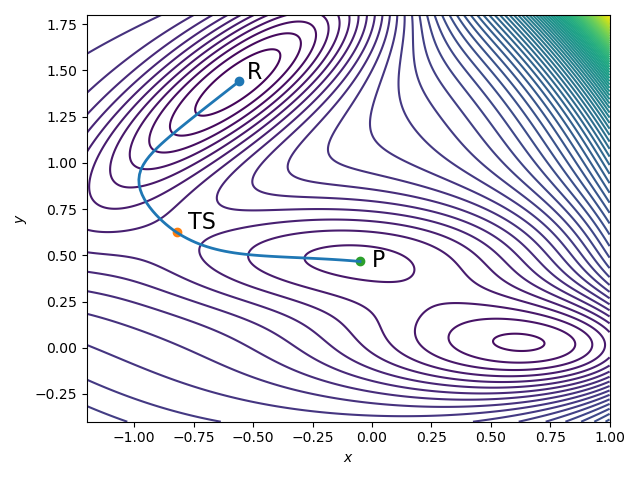
Mathematically, we can represent a chemical reaction as a transition path through configuration space, $\mathbb{R}^{N \times 3}$, where $N$ is the number of atoms in the molecule. Figure 1 illustrates such a transition path. The molecule starts in the reactant state, labeled $R$. Because a chemical reaction occurs only when sufficient energy is supplied to the system, the reactant lives near a local minimum of the energy landscape. Figure 1 is a contour diagram: the curved lines indicate level sets of equal energy, and where the contours are closer together, the local energy profile is steeper. Local minima, such as the reactant and product states, therefore appear as flat basins.
As the reaction progresses, the system climbs out of the reactant basin and moves uphill in energy until it reaches the transition state, labeled $TS$. After crossing this energy barrier, it relaxes downhill again into the product configuration, labeled $P$. A central task in computational chemistry is to calculate the full transition path from $R$ to $P$ that passes through the transition state. This is important to fully characterize the chemical reaction, i.e. to know which bonds break, how the atoms move and electrons rearrange, and to calculate the exact activation energy required to make the reaction happen.
Calculating Transition Paths
For these reasons, the calculation of transition paths from reactants, over the transition state, to products has received considerable attention in the computational chemistry literature. Over the years, several famous methods have been developed, among which the nudged elastic band (NEB) and string methods are perhaps the best known. Both methods aim to compute the full transition path $x(s)$ with $x(0)=R$ and $x(1)=P$, where $s \in [0,1]$ denotes a normalized path parameter, by placing a number of molecular instances or \textit{images} along the path. They rely on the key property that the transition path is locally parallel to the energy gradient:
\[\frac{dx(s)}{ds} \parallel \nabla E(x(s)). \tag{1}\]The function $E(x)$ is the potential energy surface (PES), and Figure 1 is, in fact, a contour plot of this surface.
Simply placing a few images on the energy landscape and moving them according to the force does not work well. Each image tends to slide downhill toward either the reactant or product minimum, rather than staying spread out along the reaction path. As a result, the intermediate images collapse into the wells and fail to capture the transition region. The NEB and String methods are designed to prevent this. In the NEB method, neighboring images are connected by artificial springs. These springs do not represent physical forces in the molecule; they are only introduced to keep the images distributed along the path. If $x_i = x(s_i)$ denotes the images along the band, then the total NEB force
\[F_i^{\mathrm{NEB}} = -\nabla E(x_i)^{\perp} + k\bigl(\|x_{i+1}-x_i\|-\|x_i-x_{i-1}\|\bigr)\tau_i,\]consists of two parts: the perpendicular component of the true force from the energy landscape $-\nabla E(x_i)^{\perp}$, which pushes the images toward the reaction path (but not downhill), and the spring force, which keeps neighboring images from bunching together. The second term above is the spring force with $\tau_i$ the local tangent to the transition path. On the other hand, the string method lets the particles evolve naturally, but reparametrizes the trajectory every few steps. The string method is slightly simpler because it doesn’t use hyperparameters like the spring constant $k$ and is, in my opinion, easier to implement.
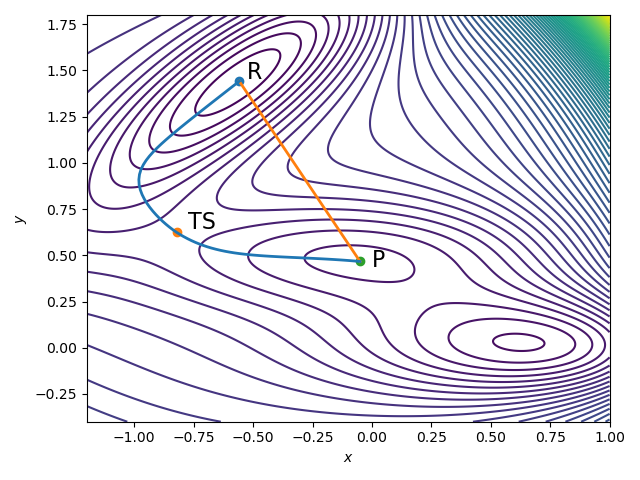
However, both methods require an initial guess for the transition path, i.e., an initial distribution of the images $x_i = x(s_i)$. In practice, the most reasonable default choice is linear interpolation between reactants and products
\[x(s_i) = (1-s_i) \text{R} + s_i \text{P}.\]Looking at Figure 2, the actual transition path is clearly not a straight line - it bends and curves trying to stay parallel to the energy gradient (1). Most chemical reactions do not happen in a straight line. As atoms move, their bonds rotate, torsion angles change and the global molecular structure adapts to a completely different conformation. Nothing about a chemical reaction is linear, but without prior knowledge of the PES, linear interpolation between reactants and products is the best initial guess one can make.
Especially in larger molecules or complex reactions, the NEB and string methods tend to suffer from poor initialization, meaning they require many iterations before converging to a reasonable approximation of the transition path. Can we do better?
Learning the Transition Path
The last three years have seen the emergence of a new research direction: building machine learning models that generate the transition path directly. The central idea is to learn from trajectories of known chemical reactions in known molecules, and to construct neural network architectures that can generalize to unseen molecules and unseen reactions. A particularly appealing approach is to use generative AI, especially diffusion models. Diffusion models are a natural fit because the transition path is not unique: thermal fluctuations cause atoms to wiggle randomly and can alter the way a reaction proceeds, even when the reactant and product states are fixed. I wrote more broadly about settings where diffusion models can create real value in Generative AI for Computational Science: A Viable Path.
Learning transition paths is more subtle than simply feeding transition-state data into a generic neural network and hoping that training converges and generalizes. Molecules are inherently three-dimensional objects, and chemistry does not change when a molecule is translated or rotated. Any useful machine learning model for this problem should therefore respect the symmetries of the underlying physics. This is not optional: rotational and translational equivariance are essential.
Over the last two weeks, I have been reading more deeply into this topic. The remainder of this post is a brief literature overview of what I found most interesting.
Learning transition paths is slightly more complicated than throwing a vanilla neural network at given transition state data and hoping training converges. Molecules are inherently three-dimensional objects, and chemistry and physics don’t change when the molecule is translated or rotated. These ML models should respect the geometric symmetries of the problem. This property is not optional: rotational and translational equivariance are a necessity.
I’ve been reading more into this topic over the last two weeks, the remainder of this post is a summary of what I’ve been reading.
-
The starting point for much of this literature is
\[\mathrm{NN}_{\theta}(Rx + t) = R\,\mathrm{NN}_{\theta}(x) + t,\]e3nn, short for Euclidean neural networks. Here, $E(3)$ denotes the group of translations, rotations, and reflections in three-dimensional Euclidean space. The key idea is to build neural network layers that are equivariant under these transformations, so that rotating or translating a molecule produces a correspondingly rotated or translated output. Mathematically, equivariance meansfor every rotation or reflection $R \in \mathbb{R}^{3 \times 3}$ and translation $t \in \mathbb{R}^3$. The
e3nnframework provides a general toolbox of equivariant building blocks, including tensor products and spherical harmonics, and many of the more specialized models below are built on top of it. -
3DReact is an end-to-end neural network that can predict the activation energy barrier, in particular the Gibbs free energy, directly from reactant and product information. The starting points are distance-based graphs representing the reactants and products, where all atoms within a certain cutoff distance are considered bonded. The distance-based graph also contains all necessary chemical information of each atom such as the valence structure, hybridization, (formal) charge, chirality, the number of connected hydrogen atoms and more. This distance-based graph then passes through e3nn to predict the activation. I loved reading this paper because it is concisely written and to the point. The work also presents a real breakthrough.
-
A related idea appears in “Diffusion-based generative AI for exploring transition states from 2D molecular graphs” - is to predict the transition state and activation energy directly from the two-dimensional graph representation of reactants and products, also known as SMILES strings. Their architecture,
TSDiff, uses graph neural network layers in a score-matched diffusion model to predict the three-dimensional configuration of the transition state. -
In “Equivariant Neural Diffusion for Molecule Generation” the authors train a diffusion model with equivariant layers for molecular generation given chemical composition and substructure constraints - a conditional diffusion model. This paper presents a new forward SDE process that also contains learnable parameters, providing better molecular generation results. This paper is very math-heavy, but the math is not very focused on molecular structures. Worth reading if you want to learn more about diffusion models!
-
“Generative Model for Constructing Reaction Path from Initial to Final States” takes diffusion models further by generating the full transition path between reactants and product, and finding the transition state as the point of highest energy along the trajectory. Their architecture adds equivariant self-attention layers to
e3nn- a beautiful connection between Transformers and computational chemistry! -
Finally, “Machine Learning Transition State Geometries and Applications in Reaction Property Prediction” produces an easy-to-read overview and comparison of several machine learning approaches for predicting transition states of reactions, including some of the methods mentioned here. I found this article a particular nice read in preparation for this post.
Let me know if you find posts like this interesting. The purpose of this channel is to follow my thinking in real time as I read papers, build models, and code through new ideas. Computational chemistry is one of the fields I find most exciting, both for its intellectual depth and for the enormous promise it holds. Stay tuned!